
A crisis overcome
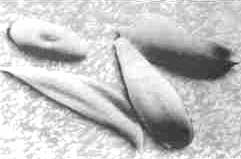 FOR the first time since modern medical practice tried to come to grips with the problem of sickle cell anaemia, a ray of hope has appeared for persons suffering from this inherited genetic disorder of the blood. The disease manifests itself by sudden and excruciating bouts of pain, referred to as 'sickle cell crises'. Scientists have now found a more cost- effective method in hydroxyurea, a cytotoxic (toxic to cells) drug, of getting relief from the pain. Hydroxyurea is reported to be the first drug to reduce the frequency of bouts of pain by almost 50 per cent.
FOR the first time since modern medical practice tried to come to grips with the problem of sickle cell anaemia, a ray of hope has appeared for persons suffering from this inherited genetic disorder of the blood. The disease manifests itself by sudden and excruciating bouts of pain, referred to as 'sickle cell crises'. Scientists have now found a more cost- effective method in hydroxyurea, a cytotoxic (toxic to cells) drug, of getting relief from the pain. Hydroxyurea is reported to be the first drug to reduce the frequency of bouts of pain by almost 50 per cent.
Sickle cell anaemia primarily affects people whose ancestors hail from Africa, India or West Asia. In the us, the disease mainly affects the African- Americans; about I in 12 of them carry a defective copy of a haemoglobin gene, which means that they have the 'trait' for the disease (but do not suffer from it). When 2 carriers of the trait marry, there is a 25 per cent chance that their child could be born with sickle cell anaernia.
The genetic defect that is responsible for the disease result4 in the production of an abnormal form of the oxygen-carrying haemoglobin. In red ing pain as well as damage to various tissues and organs. In fact, some African cultures have named the disease for its episodes of pain: in Ghana, the disease is known as Chwe chwee-chwe, meaning "relentless repetitive chewing".
In a nationwide clinical study conducted by Samuel Charache and his colleagues in the us, the efficacy.of hydroxyurea in reducing the frequency of painful crises in adults - with a history of 3 or more crises a year - was tested (New England Journal of Medicine, May 18, 1995). One hundred forty-eight men and 151 women suffering from the disease were studied. After a 4-week run-in period during which only tablets of folic acid (a nutritional supplement) were dispensed, the patients were randomly assigned to 1 of 2 groups: 'experimental' and 'controls'.
The 'experimental' group received hydroxyurea worth 15 ing per kg body weight each day to begin with, with the dosage being increased by 5 mg per kg every 12 weeks. The other group - the 'controls' - received starch tablets as a placebo. Blood samples were analysed every 2 weeks and the treatment was stopped if there were untoward signs, such as a severely depressed haernoglobin level.
The 152 patients who were put on hydroxyurea reported markedly lower frequencies of crises than those who were given the placebo. Also, they had less need of blood transfusions. The trial was stopped after an average follow-up time of 21 months.
Earlier, Orah S. Platt of Boston Children's Hospital had studied the natural history of the sickle cell disease in 3,578 pati@nts taken from a wide cross section of people. The results of the study, also published in blood cells, this abnormal haemoglobin forms rod-like structures that lead to an elongation and stiffening of the cell into a characteristic crescent or sickle-like shape from which the disease gets its name. various tissues Sickled red blood cells are neither supple nor spherical; this makes it very difficult for them to squeeze through narrow blood vessels, which get blocked as a result. The block- age starves different parts of the body of oxygen and causes periodic, self-limit- the New England Journal of Medicine (in 1991), had pointed out that the 'pain rate'- number of painful episodes per year - was a measure of the clinical severity of the disease and correlated with early death in patients over the age of 20 years. Fatality in the disease results on account of enlargement of the heart, liver and kidneys, bone damage and stroke. Apart from the body, the disease can also scar the mind, lead to a disruption of lifestyles, and hamper psychological growth of children. Hydroxyurea reduces this psychosocial fallout and offers patients the possibility of leading normal productive lives.
The secret of hydroxyurea's success appears to lie in the nature of haernoglobin itself. Haemoglobin is a protein made up to 2 copies each of 2 different subunits. Each of these subunits is encoded by a different gene. In adults, the genes (and the subunits) are called alpha and beta; in foetuses, haemoglobin is made up of 2 subunits-encoded by the alpha gene and 2 encoded by a different gene called gamma. In the course of the transition fron!i the embryo to the adult stage, the gamma gene 'switches off and the beta gene 'switches on'; in consequence, the foetal form of haemoglobin gives way to the adult form.
It has been known since the early 1950s that foetal haemoglobin has a protective effect on the polymerisation of the deoxygenated form of sickle cell haemoglobin, and the effect is greater than that caused by comparable amounts of (normal) adult haemoglobin. In certain individuals, there is a hereditary persistence of foetal haemoglobin because the gamma chain is not fully switched off. Communities in which sickle cell disease is associated with a marked elevation of foetal haemoglobin - such as those in central and southern India and the eastern provinces of Saudi Arabia - betray a reduced level of anaernia and generally mild clinical manifestations of the disease (however, high levels of foetal haemoglobin are not universally protective).
These observations had generated the possibility of awakening foetal haemoglobin, as- it were, as a therapy for sickle cell anaemia. Considerable interest was aroused, several years ago, when the administration of a drug called 5- azacytidine - a cytotoxin - to adult baboons stimulated the production of their foetal haemoglobin; other cytotoxic compounds too turned out to have a similar effect. Hydroxyurea appeared to be the most suited for clinical trials because of the relative ease of its administration and its safety. By means as yet unknown, hydroxyurea appears to ,switch on' the haemoglobin gamma gene whose functioning is necessary for the production of foetal haernoglobin.
One should bear in mind that adverse effects of longterm use of hydroxyurea are still unclear (depressed bone marrow fiinctioning is a possibility), for which reason it is still not being recommended for use by children and pregnant women. However, there is little doubt about the fact that its use can make sickle cell anaemia treatment easier and less extensive.
Related Content
- COVID-19 in North and Central Asia: impacts, responses and strategies to build back better
- Philippines Digital Economy Report 2020
- Community forest rights and the pandemic: Gram Sabhas lead the way
- A future for the world’s children? A WHO-UNICEF-Lancet
- Edible insects could be a key ingredient to avoiding a global food crisis, researchers insist
- Wealthy but unhealthy: overweight and obesity in Asia and the Pacific- trends, costs, and policies for better health